
More Information
Submitted: 28 October 2020 | Approved: 07 December 2020 | Published: 08 December 2020
How to cite this article: Carlone G, Prisco A, Vittoria F, Barbi E, Carbone M. A girl with a stiff neck. J Adv Pediatr Child Health. 2020; 3: 058-060.
DOI: 10.29328/journal.japch.1001019
ORCiD iD: orcid.org/0000-0002-2926-5462
Copyright License: © 2020 Carlone G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Xanthogranuloma; Juvenile; Central nervous system; Systemic; Imaging

A girl with a stiff neck
G Carlone1*, A Prisco2, F Vittoria3, E Barbi1,4 and M Carbone3
1Department of Medicine, Surgery and Health Sciences, University of Trieste, Trieste, Italy
2Department of Medicine, Surgery and Health Sciences, University of Campania Luigi Vanvitelli, Caserta, Italy
3Orthopedics and Traumatology Unit, Institute for Maternal and Child Health, IRCCS, Burlo Garofolo, Trieste, Italy
4Department of Paediatrics, Institute for Maternal and Child Health, IRCCS, Burlo Garofolo, Trieste, Italy
*Address for Correspondence: G Carlone, Department of Medicine, Surgery and Heath Sciences, University of Trieste, Trieste, Italy, Tel: +39 3280234342; Email: [email protected]
Juvenile xanthogranuloma (JXG) is a rare form of non-Langherans cell histiocytosis (non-LCH) observed almost exclusively in infants and young children. It is rarely systemic, involving extracutaneous sites, such as the liver, lungs, spleen, kidney, pancreas, bone or central nervous system. Systemic JXG may be associated with significant complications requiring aggressive medical or surgery care; especially, central nervous system lesions are difficult to treat and reported to be possibly fatal. Clinical presentation of JXG of central nervous system is not specific and is related to the involved site while magnetic resonance imaging examination remains the first choice for localizing the lesions. If no other system is involved, surgical excision could be sufficient.
A five-year-old girl was admitted for a stiff neck, progressively started since two months, associated with right shoulder pain. Her medical history was unremarkable.
Physical examination was remarkable for cervical muscle stiffness, right head deviation, symmetric upper limbs distal loss of strength with normal reflexes. Pain scored seven according to a Visual Analogic Scale.
Blood tests showed normal blood cells count, C reactive protein and erythro-sedimentation rate (ERS). A brain and cervical magnetic resonance (MRI) was performed (Figures 1,2).
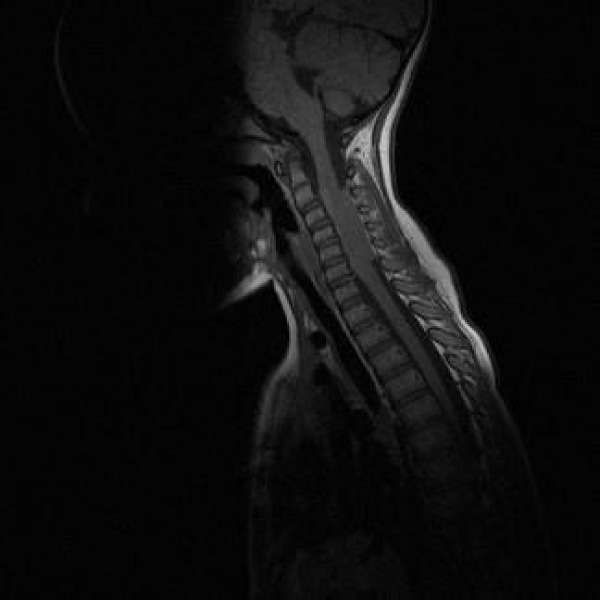
Figure 1: MRI 1
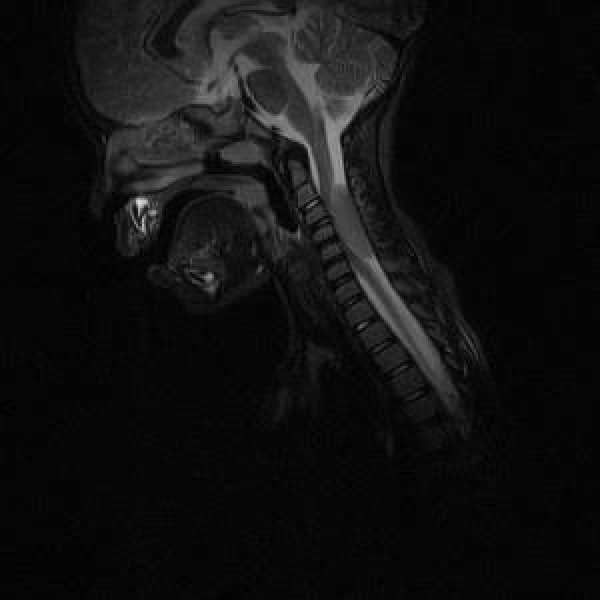
Figure 2: MRI 2
Which is the most likely diagnosis?
a) Primitive neuroectodermal tumour (PNET) of the central nervous system
b) Meningioma
c) Xanthogranuloma
d) Langherans cell histiocytosis
e) Spondylodiscitis
The answer is C
Juvenile xanthogranuloma (JXG) is a rare form of non-Langherans cell histiocytosis (non-LCH) observed almost exclusively in infants and young children [1,2]. It usually presents as an orangeyellowish colour spontaneously regressing single or multiple, 1-20 mm in size, nodular, well demarked cutaneous lesion on the trunk, scalp, face or extremities. JXG is rarely systemic, involving extracutaneous sites, such as the liver, lungs, spleen, kidney, pancreas, bone or central nervous system [2,3]. JXG has been associated with juvenile chronic myelogenous leukemia, neurofibromatosis type 1, urticaria pigmentosa, insulin-dependent diabetes mellitus, aquagenic pruritis and cytomegalovirus infection [4]. In contrast to the cutaneous form, systemic JXG may be associated with significant complications requiring aggressive medical or surgery care. Central nervous system lesions are generally difficult to treat and reported to be possibly fatal [4,5]. The frequency of systemic involvement has been estimated to be 5% - 10%, with 1% - 2% of patients showing central nervous system involvement, which can be classified according to its localization as cerebral intraparenchymal, dural and cranial or spinal nerve derived [2,5].
The patient’s MRI is suggestive of JXG showing an isointense lesion on T1 and T2-weighted imaging, homogeneously enhanced after administration of Gadolinium. No perilesional oedema, calcifications, bleeding or vascular malformation were revealed by SWI [6]. MRI imaging rules out PNET which typically shows isointense lesions on T1 and T2-weighted sequences, with heterogenous enhancement after administration of Gadolinium [7]. Meningiomas are usually asymptomatic and discovered incidentally on imaging with a typical MRI signal that consists of hypointensity on the T1-weighted and isointensity to slight hyperintensity on the T2 sequence, with avid homogeneous enhancement after contrast. Most of these lesions shows a characteristic peripheral dural thickening called tail sign [8]. LCH, which usually presents as diabetes insipidus secondary to pituitary involvement, is characterized by hypointense nodular lesions on T1-weighted and hyperintensity on T2 signal [9].
Finally, spondylodiscitis is more common in children younger than three years and in this clinical condition ESR is almost always elevated [10]. It can be diagnosed in presence of reduced disc height, disc hypointensity on T1-weighted sequence, disc hyperintensity on T2-weighted MRI image, or disc enhancement [11].
How is this diagnosis confirmed?
a) Biopsy
b) MRI spectroscopy
c) Clinical features
d) Blood laboratory tests
e) b + c
The answer is A
Biopsy and pathological examination remain the gold standard for the diagnosis of JXG involving central nervous system. Immunohistochemical studies demonstrates diffuse reactivity for CD68+ and a large amounts of foamy cells, giant Touton-type cells, with negative CD1a or S100, which rule out a LCH .
Clinical presentation is not specific and is related to the involved site. While MRI imaging characteristics are not specific and variable, spectroscopy has not been reported to be useful, and MRI examination remains the first choice for localizing the lesions. JXG is not associated with serum lipid abnormalities [12].
What is the best management approach?
a) Surgery
b) Chemotherapy
c) Radiation therapy
d) Watchful waiting
a) b + c
The answer is A and D
Complete resection of well-defined lesion can be curative. Indeed, in this patient an open-door laminoplasty was performed, as a respectful approach for the anatomy of the growing spinal column. When surgery is not feasible (in patients with inaccessible lesions or diffuse visceral infiltration), corticosteroids, chemotherapy, radiation or a combination treatment are all reported to be effective [2]. The main drugs used are cyclosporine, vinca alkaloids, etoposide and methotrexate [3].
JXG of central nervous system can be a primary lesion or a part of a widespread disease, so when it occurs a thorough search for other sites of involvement must be undertaken. If no other system is involved, data available in literature, suggest that surgical excision is sufficient and 12-months follow-up demonstrates no recurrency of disease [13].
Patient outcome
The patient clinically improved after surgery without any relapse after 12-months of follow-up. However, the patient’s follow-up still lasts in order to rule out development of cervical kyphosis that could be a fearsome complication related to posterior surgery cervical spine.
- AlQazlan S, Albakr A, Al Towim A, Alsaadan Y, Hassan H, et al. Aggressive surgical management of recurrent intracranial juvenile xanthogranuloma: case report and review of the literature. Childs Nev Syst. 2020; 36: 213-217. PubMed: https://pubmed.ncbi.nlm.nih.gov/31385088/
- Wang B, Jin H, Zhao Y, Ma J. The Clinical Diagnosis and Management Options for Intracranial Juvenile Xanthogranuloma in Children: Based on Four Cases and Another 39 Patients in the Literature. Acta Neurochir. 2016; 158: 1289-1297. PubMed: https://pubmed.ncbi.nlm.nih.gov/27173098/
- Orsey A, Paessler M, Lange BJ, Nichols KE. Central Nervous System Juvenile Xanthogranuloma with Malignant Transformation. Pediatr Blood Cancer. 2008; 50: 927-930. PubMed: https://pubmed.ncbi.nlm.nih.gov/17520744/
- Freyer DR, Kennedy R, Bostrom BC, Kohut G, Dehner LP. Juvenile xanthogranuloma: Forms of systemic disease and their clinical implications. J Pediatr. 1996; 129: 227-237. PubMed: https://pubmed.ncbi.nlm.nih.gov/8765620/
- Rampini PM, Alimehmeti RH, Egidi MG, Zavanone ML, Bauer D, et al. Isolated Cervical Juvenile Xanthogranuloma in Childhood. Spine. 2001; 26: 1392-1395. PubMed: https://pubmed.ncbi.nlm.nih.gov/11426158/
- Jingkai S, Ni C, Qiang Y, Wang W, Zhang S, et al. Multimodel MRI features of an intracranial juvenile Xanthogranuloma. Child's Nervous System. 2019; 35: 871–887. PubMed: https://pubmed.ncbi.nlm.nih.gov/30820641/
- Velivela K, Rajesh A, Uppin MS, Purohit AK. Primary Intracranial Peripheral PNET. A Case Report and Review. Neurol India. 2014; 62: 669-673. PubMed: https://pubmed.ncbi.nlm.nih.gov/25591683/
- Vernooij MW, Ikram MA, Tanghe HL, Vincent AJPE, Hofman A, et al. Incidental Findings on Brain MRI in the General Population. N Engl J Med. 2007; 357: 1821-1828. PubMed: https://pubmed.ncbi.nlm.nih.gov/17978290/
- De La Hoz Polo M, Rebollo Polo M, Fons Estupiña C, Muchart López J, Cruz Martinez O, et al. Neuroimaging of Langerhans Cell Histiocytosis in the Central Nervous System of Children. Radiologia. 2015; 57: 123-130. PubMed: https://pubmed.ncbi.nlm.nih.gov/24837565/
- Cushing AH. Diskitis in Children. Clinical Infectious Diseases. 1993; 17: 1–6. PubMed: https://pubmed.ncbi.nlm.nih.gov/8353227/
- Principi N, Esposito S. Infectious Discitis and Spondylodiscitis in Children. Int J Mol Sci. 2016; 17: 539. PubMed: https://pubmed.ncbi.nlm.nih.gov/27070599/
- Guillou L, Folpe AL. Bone and Soft Tissue Pathology (E-Book), 2010.
- Fulkerson DH, Luerssen TG, Hattab EM, Kim DL, Smith JL. Long-Term Follow-Up of Solitary Intracerebral Juvenile Xanthogranuloma. Case report and Review of the Literature. Pediatr Neurosurg. 2008; 44: 480-485. PubMed: https://pubmed.ncbi.nlm.nih.gov/19066439/